

Yo sospecho, sospecho siempre.Umberto Eco
CLN2: lo que la epilepsia nos hace perder de vista
En su obra El libro de oro, el filósofo y escritor romano Lucio Anneo Séneca afirma: “No puede el médico curar bien sin tener presente al enfermo”.1 Esta premisa, tangencial para el ejercicio de la medicina, cobra mayor valor cuando de enfermedades neurológicas se trata, como las encefalopatías epilépticas o enfermedades neurodegenerativas, entre las que destaca la lipofuscinosis neuronal ceroide tipo 2 (CLN2; también conocida como lipofuscinosis ceroide neuronal infantil tardía, enfermedad de Batten o enfermedad de Jansky-Bielschowsky),2 una enfermedad rara, que consiste en un trastorno neurodegenerativo por depósito lisosomal, autosómico recesivo, que inicia desde el nacimiento, se manifiesta en los primeros años de vida y es causado por la actividad deficiente de la enzima tripeptidil-peptidasa 1 (TPP1).3-5 La CLN2 de presentación clásica cursa con inicio de convulsiones entre los 2 y los 4 años, seguido de deterioro cognitivo (retraso o involución del lenguaje), pérdida visual y atrofia cerebral, o una combinación de ellos, que generalmente resultan en la muerte del paciente a una edad temprana, entre los 8 y los 15 años.4 Por tanto, es imperativo que el médico de consulta general y el pediatra tengan el radar activo en todo momento para la detección oportuna de pacientes con CLN2 (Figura 1).6

El fenotipo CLN2 es la forma más común de las lipofuscinosis neuronales ceroides (CLN),3 una familia de trastornos neurodegenerativos de rápida evolución.7
Las CLN son la principal causa de demencia en niños y adolescentes y se caracterizan por la acumulación de lipopigmento autofluorescente (lipofuscina ceroide) en las neuronas, la retina y otros órganos.7 Su presentación varía según la edad, los síntomas y la evolución de la enfermedad, y tradicionalmente han sido identificadas por una triada de síntomas incapacitantes, que incluyen crisis epilépticas, demencia y pérdida de la visión.7 Inicialmente, las CLN fueron clasificadas de acuerdo con la edad de aparición de los síntomas. Gracias a la creciente identificación de los defectos genéticos subyacentes, se han desarrollado nuevas nomenclaturas y esquemas de clasificación.8
Infortunadamente, debido a que la CLN2 presenta una evolución clínica similar a la de otros trastornos convulsivos y neurodegenerativos, su detección solo tiene lugar después de varias sospechas diferentes al diagnóstico definitivo, cuando la enfermedad ya se encuentra en un estado avanzado.3
Es cierto que la epilepsia en la población pediátrica es común y tiene un amplio espectro de manifestaciones clínicas y de diferentes causas,9 aunque solo la minoría de estas tiene un tratamiento dirigido a evitar la causa subyacente, razón suficiente para que la CLN2 siempre sea una de las primeras sospechas diagnósticas que deba descartarse en todo paciente pediátrico que presente convulsiones no provocadas de inicio reciente o que empeoran, conducta que se hace obligatoria cuando, además de las convulsiones, el paciente exhibe un cuadro clínico de ataxia (incoordinación del movimiento voluntario), retraso o involución del lenguaje o pérdida de la visión, síntomas que pueden aparecer simultánea o progresivamente en el curso de la enfermedad.3
Para evitar los retrasos en la detección oportuna de la CLN2 y, por tanto, el avance de la enfermedad hacia un punto de no retorno donde el daño sea irreversible,5 los expertos recomiendan la remisión inmediata al diagnóstico de laboratorio de todo paciente pediátrico con sospecha de CLN2 en la primera consulta,3 incluso si este no muestra todo el cuadro clínico característico de este trastorno.
A continuación, se describe el algoritmo diagnóstico que los médicos de consulta general y pediatras deben seguir en estos casos.
Con el radar activo para que la CLN2 no dé un paso más
La prueba de referencia para el diagnóstico de laboratorio de la CLN2 es el tamizaje bioquímico y, en caso de dar positivo, la confirmación mediante análisis molecular, con los que se busca demostrar la actividad deficiente de la enzima TPP1, que es medida, generalmente, en papel filtro (gotas de sangre seca) y también es posible en leucocitos o fibroblastos (siendo esta muestra biológica confirmatoria), y la identificación de las mutaciones patogénicas en cada alelo del gen TPP1/CLN2, respectivamente (Figura 2).3 En Colombia, el Laboratorio Clínico Biolab, como parte del proyecto Epilepsia y Genética, se encarga de la recolección y el procesamiento de la muestra, de forma gratuita, para el diagnóstico temprano de la CLN2. Para más información al respecto, puede contactarse llamando al número 301 679 9960 o escribiendo al correo electrónico info@laboratorioclinicobiolab.com
CLN2: lipofuscinosis neuronal ceroide tipo 2; EEG: electroencefalograma; ERG: electrorretinografía; FEI: fotoestimulación intermitente; PVE: potenciales visuales evocados; RM: resonancia magnética; TPP1: tripeptidil-peptidasa 1.
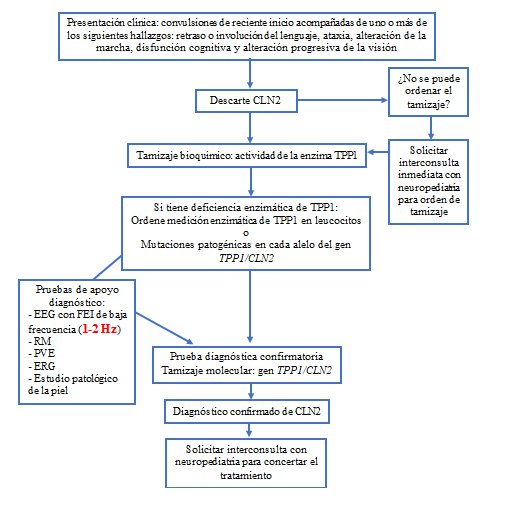
La enzima TPP1 es una exopeptidasa serina, con óptima actividad in vitro en pH ácido.3 Otra opción válida para el diagnóstico temprano es el tamizaje molecular mediante un panel de epilepsia. Existen diferentes paneles ofrecidos en Colombia y varían dependiendo de la cantidad de genes incluidos. Las ventajas del tamizaje molecular son que permite ampliar el potencial de investigación de la etiología genética de las crisis epilépticas y determina las posibles implicaciones para el cuidado de los pacientes.7 Con una sola prueba de tamizaje se puede confirmar la presencia de 40 genes o más relacionados con otros trastornos neurodegenerativos, incluida la CLN2.7
Otros signos clínicos que aumentan la sospecha de CLN2: que el radar continúe activo
Electroencefalograma con fotoestimulación intermitente de baja frecuencia
A menudo, el electroencefalograma (EEG) constituye una de las primeras pruebas diagnósticas complementarias que se realizan en casi todos los pacientes pediátricos que consultan por convulsiones, independientemente del nivel de sospecha. En el caso de la CLN2, el EEG puede detectar actividad irritativa, ralentización de la actividad de fondo y alteraciones epileptiformes en las regiones posteriores.3
Uno de los grandes problemas de los protocolos de EEG en los diferentes centros en Colombia para llegar a un diagnóstico temprano de CLN2 es la no utilización de fotoestimulación que inicie con bajas frecuencias (1-2 Hz). El EEG con fotoestimulación intermitente (FEI), realizado en una frecuencia de 1 a 2 Hz, es un procedimiento de activación de la actividad epileptiforme. En gran parte de los EEG de los pacientes diagnosticados con CLN2 se ha reportado una respuesta positiva con los destellos de baja frecuencia. Cabe mencionar que si la FEI es llevada a cabo en frecuencias mayores de 2 Hz, hay riesgo de que no se genere una respuesta positiva, el enfoque diagnóstico cambie y el paciente pierda meses que pueden significar un deterioro considerable de sus habilidades, como caminar, o del lenguaje. En la fase temprana de la enfermedad, puede no darse una respuesta positiva a la fotoestimulación; sin embargo, esto no descarta que el paciente presente CLN2.3, 4
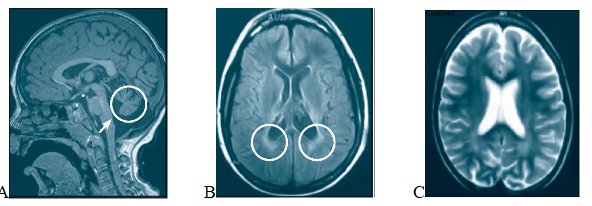
Resonancia magnética
Los hallazgos mostrados por los métodos imagenológicos, como la resonancia magnética (RM) cerebral, incluyen atrofia cerebelar y cortical, disminución en el volumen de la sustancia gris e hiperintensidades periventriculares en la sustancia blanca (Figura 3),3, 7 las cuales constituyen datos diagnósticos importantes que no deben interpretarse como leucodistrofia.3
Diagnóstico visual
En pacientes con CLN2, la pérdida de la visión tiene una presentación tardía en el curso de la enfermedad;2 es decir, después de los 4-5 años, cuando los pacientes quedan en silla de ruedas, antes de morir (entre los 8 y 12 años).10
La pérdida de la visión en la CLN2 ocurre secundaria a una degeneración progresiva de la retina de fisiopatología incierta.2 Las manifestaciones oftalmológicas de la CLN2 están íntimamente relacionadas con el grado de función neurológica y la edad del paciente e incluyen: maculopatía en ojo de buey, atrofia del epitelio pigmentario de la retina, anomalías en la autofluorescencia del fondo ocular, trastornos de la pigmentación periférica y una electrorretinografía reducida o nula.2
Pronóstico de la CLN2: razones para no bajar la guardia
La CLN2 es una enfermedad rara y, a la fecha, no se cuenta con una incidencia estimada para Colombia, por lo que se toman datos provenientes de otros países. Por ejemplo, en Alemania Occidental presenta una incidencia de 0,46 por cada 100.000 nacimientos vivos y en el norte de Europa registra una prevalencia estimada de 0,6 a 0,7 por cada 100.000 nacimientos vivos.4 Como se ha informado en otros reportes, el período de latencia para el diagnóstico es significativo. La CLN2 es diagnosticada después de una duración promedio de 22,7 meses (1,8 años) desde el inicio de los síntomas y después de haberse producido un deterioro funcional importante.10 La demora entre el inicio de los síntomas y el diagnóstico representa, por tanto, una carga considerable para los pacientes con CLN2 y sus familias, debido a que retrasa el asesoramiento genético, aumenta el riesgo de tener hermanos que puedan verse afectados por la enfermedad y retarda el inicio de las estrategias de manejo clínico y psicosocial.11 En consecuencia, en vista de que los pacientes con CLN2 presentan un rápido deterioro en la función clínica (durante un período de 2 a 3 años) desde la aparición de los primeros síntomas hasta las etapas finales de la enfermedad, donde se observa un deterioro funcional casi total, el pronóstico de la CLN2 es devastador (Figura 4 y Tabla 1).6, 7, 11, 12
(Textos de la figura: Suma de los puntajes en los dominios motor y lenguaje IC 95% del muestreo Media estimada Percentil 10 Percentil 25 Percentil 75 Percentil 90 Edad (meses))

Tabla 1. Lipofuscinosis neuronal ceroide: fenotipos, genes mutados, edad de inicio de la enfermedad y cuadro clínico
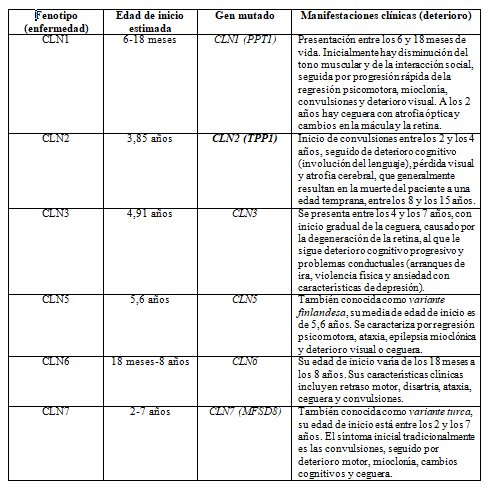
En Latinoamérica, aunque se creía que el fenotipo más común de LCN es la CLN2 infantil tardía, también denominada clásica, caracterizada por una edad de inicio temprana (de 2 a 4,5 años), encefalopatía progresiva y pérdida de la visión,7, 13 recientemente se ha identificado fenotipos atípicos que son más frecuentes en la población latinoamericana, con respecto a otras poblaciones en el mundo, y que se diferencian de la forma clásica en que se presentan en edades de inicio tardías (de 2,5 a 10 años) y en un curso de la enfermedad prolongado.13 Véase la Tabla 2, donde se muestran otras diferencias clave para la diferenciación e identificación oportunas de las formas clásica y atípica de la CLN2.
Tabla 2. Cuadro comparativo entre las formas clásica y atípica de la CLN2 en población latinoamericana.

Hasta octubre de 2019, año en el que se aprobó la terapia de reemplazo enzimático intraventricular con cerliponasa Alfa en Colombia, solamente se disponía de tratamiento paliativo para los pacientes con CLN2.11 Con la aprobación de la Administración de Alimentos y Medicamentos (FDA) de la terapia de reemplazo enzimático intraventricular como primera línea de tratamiento de la CLN2, el retraso en el diagnóstico indica que la evolución de la enfermedad pudo haber sido considerable antes de iniciar el tratamiento. Este retraso resulta en un deterioro casi total de las funciones motoras y del lenguaje normales. Por tanto, el diagnóstico temprano es benéfico en todos los pacientes con CLN2 en virtud de que ya existe un tratamiento aprobado, además de las terapias experimentales que están siendo desarrolladas.11
Con el radar activo, las conclusiones siempre serán positivas
Por último, a continuación, se exponen las conclusiones más relevantes y que tanto los médicos de consulta general como los pediatras deben tener en cuenta para contrarrestar el impacto devastador de la CLN2 en los pacientes que la padecen:
- El fenotipo CLN2 es la forma más común de las lipofuscinosis neuronales ceroides (LCN), una familia de trastornos neurodegenerativos pediátricos de rápida evolución y mal pronóstico.
- Los expertos recomiendan la remisión inmediata al diagnóstico de laboratorio (tamizaje bioquímico de la enzima TTP1 y posterior confirmación molecular de las mutaciones en el gen TPP1/CLN2) de todo paciente pediátrico, usualmente con inicio entre los 2 y los 9 años para evolución atípica, con el fin de descartar CLN2 desde las primeras consultas.
- El retraso en la detección oportuna de la CLN2 conduce al avance de la enfermedad hacia un punto de no retorno, donde el daño es irreversible.
- La satisfacción de cambiar el curso de una enfermedad devastadora como la CLN2 no tiene precio; por tanto, ¡mantenga el radar activo desde el primer momento!
Referencias
- Séneca LA. El libro de oro. Barcelona, España: Ediciones Brontes L.S.; 2019.
- Orlin A, Sondhi D, Witmer MT, Wessel MM, Mezey JG, Kaminsky SM, et al. Spectrum of Ocular Manifestations in CLN2-Associated Batten (Jansky-Bielschowsky) Disease Correlate with Advancing Age and Deteriorating Neurological Function. Plos ONE. 2013;8(8):e73128.
- Fietz M, AlSayed M, Burke D, Cohen-Pfeffer J, Cooper JD, et al. Diagnosis of neuronal ceroid lipofuscinosis type 2 (CLN2 disease): Expert recommendations for early detection and laboratory diagnosis. Mol Gen Metab. 2016;119(1-2):160-7.
- Pérez-Poyato M, Pineda-Marfa M, Ferrer-Abizanda I, Rodriguez-Revenga L, Cusí-Sánchez V, Martínez-González MJ, et al. Late Infantile Neuronal Ceroid Lipofuscinosis: Mutations in the CLN2 Gene and Clinical Course in Spanish Patients. J Child Neurol. 2012;28(4):470-8.
- Beltrán L, Reyes-Valenzuela G, Loos M, Vargas R, Lizama R, Spinsanti P, et al. Late-onset childhood neuronal ceroid lipofuscinosis: Early clinical and electroencephalographic markers. Epilepsy Res. 2018;144:49-52.
- BioMarin. A cuál de estos niños usted estudiaría para la enfermedad CLN2 [Internet]. 2017. Disponible en: cln2connection.com.co
- Espitia OM. CLN2: diagnóstico y tratamiento. Seminario web BioMarin; junio 11 de 2020; Bogotá, Colombia.
- Aungaroon G, Hallinan B, Jain P, Horn PS, Spaeth C, Arya R. Correlation Among Genotype, Phenotype, and Histology in Neuronal Ceroid Lipofuscinoses: An Individual Patient Data Meta-Analysis. Pediatr Neurol. 2016;60:42-48.e4.
- Modalsli K, Surén P, Lund-Søraas C, Bakken IJ, Lossius MI, Stoltenberg C, et al. Seizures, syndromes, and etiologies in childhood epilepsy: The International League Against Epilepsy 1981, 1989, and 2017 classifications used in a population-based cohort. Epilepsia. 2017;58(11):1880-91.
- Specchio N, Bellusci M, Pietrafusa N, Trivisano M, de Palma L, Vigevano F. Photosensitivity is an early marker of neuronal ceroid lipofuscinosis type 2 disease. Epilepsia. 2017;58(8):1380-8.
- Nickel M, Simonati A, Jacoby D, Lezius S, Kilian D, Van de Graaf B, et al. Disease characteristics and progression in patients with late-infantile neuronal ceroid lipofuscinosis type 2 (CLN2) disease: an observational cohort study. Lancet. 2018;2(8):582-90.
- Schulz A, Kohlschütter A, Mink J, Simonati A, Williams R. NCL diseases — clinical perspectives. Biochim Biophys Acta. 2013;1832:1801-6.
- Kohan R, Carabelos MN, Xin W, Sims K, Guelbert N, Cismondi IA, et al. Neuronal ceroid lipofuscinosis type CLN2: A new rationale for the construction of phenotypic subgroups based on a survey of 25 cases in South America. Gene. 2013;516(1):114-21.








Deja un comentario